(8) mol2chemfig でお手軽に分子を描いてみる
はじめに
chemfig で複雑な分子を書こうとするとかなり大変です。どうにか楽はできないものでしょうか。
幸いにも分子の構造に関する情報はすでにあちこちで公開されています。そのような情報を活用できればかなりお手軽に分子が描けてしまいそうな気がします。
本稿では mol2chemfig を使ってこれを実現する方法について紹介します。
やりたいことは、
- (mol2chemfig で対応しているフォーマットである) SMILES, MOL, SDF のいずれかで描きたい分子の情報を取得する
- mol2chemfig でこれを chemfig に変換する
- 変換したものをメインの
.texに入れ (もしくは\inputして) コンパイル
です。
まず SMILES, MOL, SDF についてごくごく簡単に説明しておきましょう。すでにご存知であればスキップして下さい。
SMILES と MOL/SDF フォーマット
SMILES 記法
SMILES は Simplified Molecular Input Line Entry System の略で、分子を文字列として表現する方法の一つです。 SMILES記法 (Wikipedia) なども参照して下さい。
例えば、エタノールであれば CCO と書きます。この例から分かるように、全ての H は原則として省略します。分岐(側鎖)は括弧に入れて書きます。
例えば tert-ブチルアルコールは C(C)(C)(C)O となります。この表記方法は一意ではありません。
エタノールは OCC とも書けますし、(普通はこうは書きませんが) C(O)C と書くこともできます。
環では、原子に番号を付け、同じ番号同士を繋ぎます。シクロヘキサンなら C1CCCCC1 となります。
芳香族の場合は原子は小文字で書きます。例えば、ベンゼンなら c1ccccc1 と書きます。
ただし、mol2chemfig ではこの記法は使えないらしく、 C1=CC=CC=C1 などと書く必要があります。
MOL/SDF フォーマット
これらは、各原子の三次元の位置と結合の情報を書くためのフォーマットです。 SDF (Structure Data File) は MOL のスーパーセットになっています。
詳細は省略しますが、例えばこんな感じのファイルになっています (水分子)。
h2o.mol
application information
3 2 0 0 0 0 999 V2000
-0.2600 0.7093 -0.4054 O 0 0 0 0 0 0
0.6321 0.3444 -0.5049 H 0 0 0 0 0 0
-0.8259 -0.0692 -0.2936 H 0 0 0 0 0 0
1 2 1
3 1 1
M END主要な化学関係のソフトウェアは MOL/SDF 形式でのエクスポートをサポートしているものが多いと思います。
SMILES/MOL/SDF の取得方法
これらの情報はさまざまなサイトで公開されていますが、私自身は PubChem を使うことが多いです。 PubChem では SMILES も SDF ファイルの取得もできます。
Wikipedia で化合物について調べると、たいていは SMILES での記述も掲載されていますので、これをコピー&ペーストすることもできます。 (ただし上記のように mol2chemfig では小文字を使った芳香族の記述は使えないので、その点に注意する必要があります。)
それほど複雑ではない分子ならば、自分で SMILES を書くのも一つの手です。 少なくとも chemfig を直接書くよりは楽だと思います。
mol2chemfig のインストール
では mol2chemfig をインストールしましょう。
mol2chemfig は TeX Live には含まれていないので、残念ながら自分でインストールする必要があります。 CTAN にあるもの (v1.5 (2024-12-21 現在)) は Python 2 で書かれており Python 3 では動きません。 そのため、unofficial ではありますが、
を使用するのが良いでしょう (以下では単に mol2chemfig と書きます)。 pip でインストールできるので、
pip install mol2chemfigPy3などとします1。
なお mol2chemfig のマニュアル (オリジナル) は CTAN にあります: mol2chemfig Documentation
mol2chemfig を使ってみる
mol2chemfig を使った変換
ひとまず使ってみましょう。
mol2chemfig -w --input=direct 'CCO' > ethanol-direct.tex
(-w は変換結果を \chemfig{ … } で囲むためのオプション, --input=direct は引数をそのまま化合物として解釈するためのオプション)
とすると、
\chemfig{
% 1
-[:330]% 2
-[:30,,,1]OH% 3
}
という内容のファイルが生成されます (もちろんリダイレクトしなければ、そのまま画面に表示されます)。
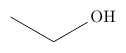
エタノールの SMILES から
chemfig
での記述に変換できました!
最後の SMILES の部分を 'OCC' や 'C(O)C' とするとどうなるかは、ご自分で確かめてみて下さい。
もちろん、SMILES そのものを書いたファイル (例えば ethanol.smi) を作成して、
echo 'CCO' > ethanol.smi # 例
mol2chemfig -w ethanol.smi > ethanol-smiles.texとする方法も良いでしょう。
便利な機能として、PubChem CID が分かっていれば、
mol2chemfig -w -y delete -i pubchem 702 > ethanol-sdf.tex
とすれば、mol2chemfig が PubChem から SDF ファイルをダウンロードして、それを
chemfig
に変換してくれます。
ここで -y delete (あるいは --hydrogens delete) は、H 原子を削除するために指定しています。
(もちろん -OH 基などの H は削除されません。)
生成したファイルを .tex で使う
あとはこれを .tex ファイルに書けば良さそうです。
ただし、mol2chemfig の出力では mol2chemfig 独自のマクロ等が使用される場合があるので、
そのために mol2chemfig.sty を読み込む必要があります。
mol2chemfig.sty も TeX Live に含まれていないので、CTAN などから取得して下さい。例えば、
wget https://mirrors.ctan.org/graphics/mol2chemfig/latex/mol2chemfig.sty
とします。今回は簡単のために mol2chemfig.sty はカレントディレクトリーに置かれているものとします。
すると、最低限必要な記述は以下のようになります。
\documentclass[10pt]{article}
\usepackage{chemfig,mol2chemfig}% mol2chemfig が chemfig を読むので、chemfig は無くてもよい
\begin{document}
% ここに mol2chemfig の出力をコピペ
% あるいは
% \input{ethanol-direct.tex}
% など
\end{document}これをコンパイルすれば、以下のような出力が得られます。
実例
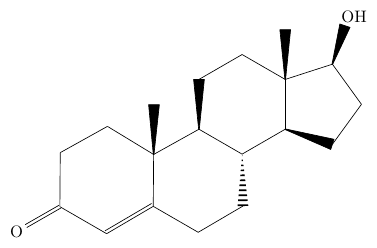
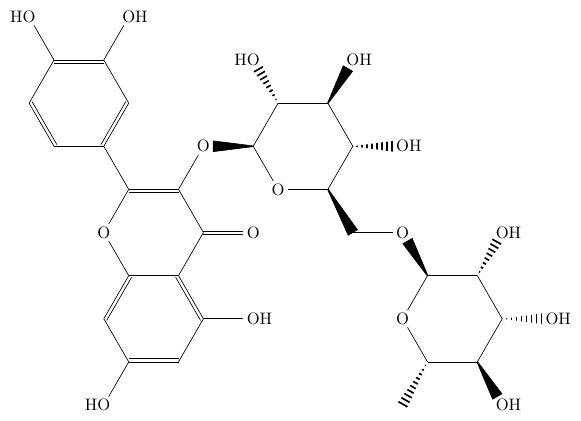
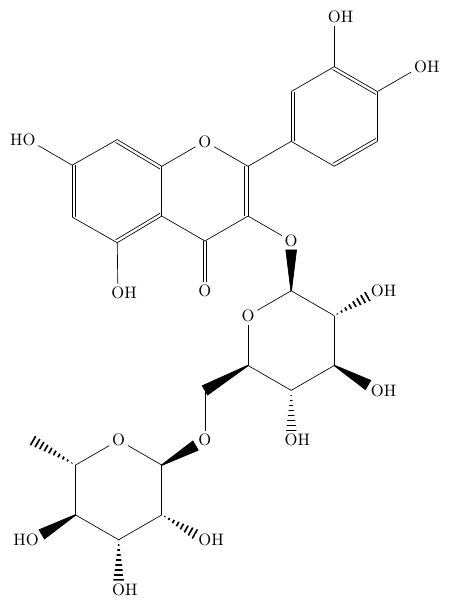
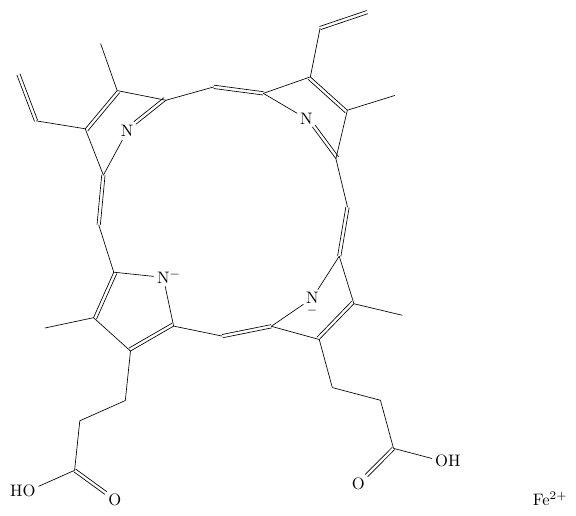
以下に PubChem から取得した SMILES (オプション: -w) 及び SDF (オプション: -w -y delete) をもとに描いた分子をいくつか、
あえて一切変更せずに載せてみます。ご覧の通り、多くの場合で分子の向き以外は SMILES でも SDF でも大差なく、特に問題なく描けています。
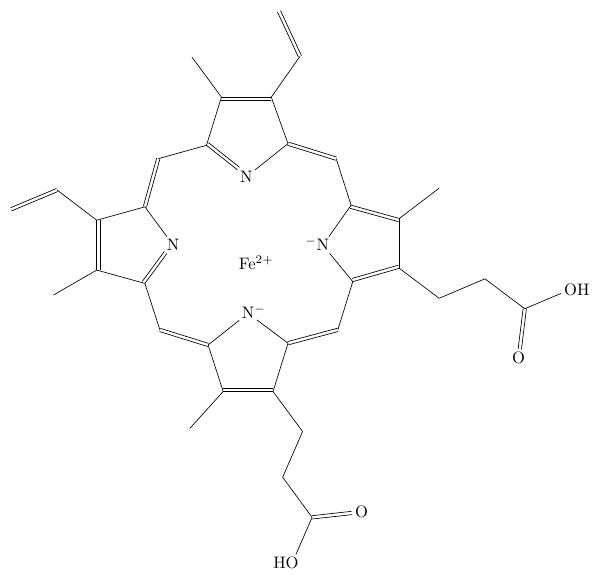
Heme b のような錯体は、SMILES をもとにしたものはかなり壊滅的で (とは言っても限られた情報でここまで再現しているのはかなり健闘していると言って良いでしょう)、 SDF をもとにしたものはかなりマシではあるものの、若干修正はしたい (私なら Fe の位置を調整して、4 つの N と線 (または点線) で結びたい) 結果となっています。 とはいえ、一から自分で書くことを考えれば、かなり省力化できるでしょう。
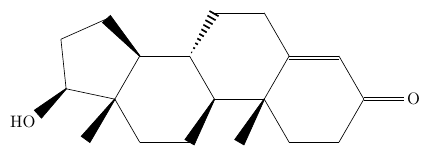
mol2chemfig の出力を手直ししてみる
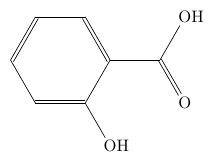
mol2chemfig の出力を使って、一部を変更した分子を描きたいかもしれません。 例として、サリチル酸の出力から一部を変更してアセチルサリチル酸を描いてみましょう。
まずは、サリチル酸の SMILES をそのまま chemfig に変換して表示してみると、次のようになります。
mol2chemfig -i direct -w 'C1=CC=C(C(=C1)C(=O)O)O'
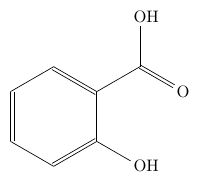
角度が気に入らないので30度回転させます。そのためには -a オプションを使うことができます。
mol2chemfig -a 30 -i direct -w 'C1=CC=C(C(=C1)C(=O)O)O'
これをベースのファイルとして使用します。
さて、-OH を「アセチル化」すれば良いことが分かりますので、この O がソースのどこにあるのかを探さねばなりません。
このくらいの小さい分子であればソースを読んで探すのもそれほど難しくはないかもしれませんが、
もっと複雑な分子の特定の炭素原子を探さねばならない状況だったとすると、それが相当困難であることは想像に難くありません。
しかしありがたいことに、mol2chemfig に -n を付けると、番号を付けてくれます。
mol2chemfig -n -a 30 -i direct -w 'C1=CC=C(C(=C1)C(=O)O)O'
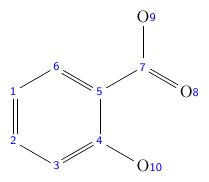
すると、今探しているのは 10 番目の原子であることが簡単に分かります。
変換されたファイルを見るとコメントで原子の番号が書かれているので、すぐ見つけることができます。
そこの OH から H を消して代わりに (-[:-90](=[:-150]O)-[:-30]) と側鎖を書けば、アセチルサリチル酸ができました!
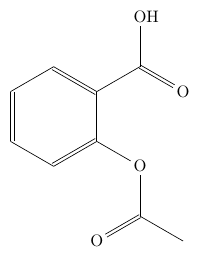
diff をとるとこんな感じです。
--- salicylicacid.tex 2024-12-16 20:11:19.313717210 +0100
+++ aspirin.tex 2024-12-16 09:55:37.421548985 +0100
@@ -7,7 +7,10 @@
-[:210]% 5
-[:270]% 4
(
- -[:330,,,1]OH% 10
+ -[:330,,,1]O% 10
+ (
+ -[:-90](=[:-150]O)-[:-30]
+ )
)
=_[:210]% 3
-[:150]% 2
手直しの程度にもよると思いますが、想像よりもかなり簡単に目的の分子を得ることができます。
chemfig
で書かれた分子を変更しようと思った場合、「対象の原子を探し出す」というのがおそらく一番難しい作業なのですが、
-n オプションとコメントのおかげでかなり効率よく変更できます。
その他の便利な機能
.tex ファイルの中から mol2chemfig を呼ぶ
コンパイル時に -shell-escape が必要となりますが、 .tex ファイルに
\mcfinput{-w ethanol.smi}と書くと、コンパイル時に mol2chemfig を実行して、その結果を入れてくれます。
\mcfinput{-w -i direct 'CCO'}なども可能です。
ただし -i pubchem を使用するとコンパイルの度に PubChem へのアクセスが発生してしまうので、
このオプションを \mcfinput で使用するのは避けるべきでしょう。
なお \mcfinput を使うとコンパイル時間が増えますし、コンパイルをする環境で mol2chemfig がインストールされている必要があります。
ですので、最終版では \mcfinput を使わずに、mol2chemfig で生成した .tex ファイルをコピー&ペースト、
もしくは \input する方が良いでしょう。
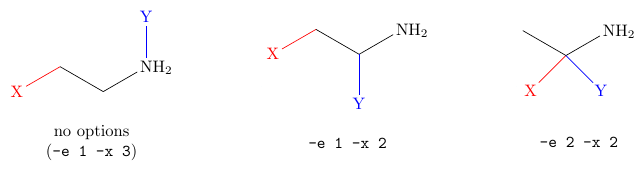
sub-molecule を定義する
sub-molecule を定義するには -l name オプションを使用します。必要に応じて -e および -x を使って、
入口原子 (entry atom)、出口原子 (exit atom) を指定します。
ここでは \mcfinput を使用しましたが、もちろんコマンドラインで mol2chemfig -l CCN -i direct "CCN" > CCN.tex
などとして .tex ファイルで \input{CCN.tex} としても良いです。
最初の例からも分かるように、水素原子の数はそのままなので、必要に応じて自分で調節しなくてはなりません。
\mcfinput{-l CCN -i direct "CCN"}
\mcfinput{-l CCNe1x2 -e 1 -x 2 -i direct "CCN"}
\mcfinput{-l CCNe2x2 -e 2 -x 2 -i direct "CCN"}
\chemfig{%
{\color{red}X}-[:30,,,,red]% X-
!{CCN}
-[:90,,,,blue]{\color{blue}Y}% -Y
}
\chemfig{%
{\color{red}X}-[:30,,,,red]% X-
!{CCNe1x2}
-[:-90,,,,blue]{\color{blue}Y}% -Y
}
\chemfig{%
{\color{red}X}-[:45,,,,red]% X-
!{CCNe2x2}
-[:-45,,,,blue]{\color{blue}Y}% -Y
}
おわりに
chemfig
は化学式等を書く際の非常に強力なツールであり、非常に綺麗な出力が得られますが、
やはり難易度が高く、他人にはおすすめしにくいというのも事実です。
私自身、他の人には「構造式エディターで描いて PDF あたりでエクスポートして \includegraphics とか使え」と言っています。
今後は、ある程度の知識がある人 (コマンド操作が問題なくできる人) には、
「構造式エディターで描いて PDF あたりでエクスポートして \includegraphics とか使うのが良いけど、
簡単なものなら mol2chemfig を使うという手もあるよ」と言うかもしれません。
何にせよ、本稿がお役に立てば幸いです。
Appendix: mol2chemfig のオプション
-
-h(Default: False) - ヘルプメッセージを表示します。
-
-b,--version(Default: False) - バージョンを表示します。
-
-i,--input(Default: file) -
引数の扱い方を指定します。
file: 引数にファイル名を取ります。direct: 引数をそのまま分子として解釈します。pubchem: 引数を PubChem での識別子と解釈します。
-
-z,--terse(Default: False) - 生成されるコードの全ての空白とコメントを削除します。
-
-r,--strict(Default: True) -
Indigo による化学構造検証を無効にします。例えば
mol2chemfig -w -i direct 'C(O)(O)(O)(O)O'はエラー (C から 5 本の結合が出ている) になりますが、
mol2chemfig -r -w -i direct 'C(O)(O)(O)(O)O'とするとエラーにならずそのまま解釈されます。 (Indigo については
https://lifescience.opensource.epam.com/indigo/index.htmlなどを参照して下さい。) -
-d,--indent(Default: 4) - インデントのスペースの数を指定します。
-
-u,--recalculate-coordinates(Default: False) - MOL/SDF に記述されている座標を破棄して、結合から原子の配置を再計算します。
-
-a,--angle(Default: 0.0) - 分子の回転角度 (反時計回り) を指定します。
-
-v,--relative-angles(Default: False) - 結合に相対角度を使います。
-
-p,--flip(Default: False) - 分子を左右反転させます。
-aと同時に指定された場合は-pが優先 (-a 60 -pも-p -a 60も反転してから 60 度回転) されるようです。 -
-q,--flop(Default: False) - 分子を上下反転させます。
-pと同時に指定することもできます。 -
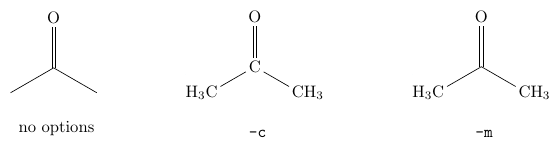
-c,--show-carbons(Default: False) - 炭素原子 (C) を表示します。
-
-m,--show-methyls(Default: False) - メチル基を表示します。
次の図は、
mol2chemfig -a -60 -w -i direct 'CC(=O)C'において-cおよび-mの有無による差を示したものです。 -
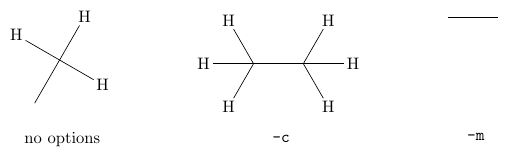
-y,--hydrogens(Default: keep) -
(陽に書かれている) 水素原子をどのように扱うかを指定します。
keep: 何もしません。add: 水素原子を追加します。delete: 水素原子を表示しません。
以下は
mol2chemfig -w -i direct 'CC([H])([H])[H]'において-y keep,-y addおよび-y deleteによる差を示したものです。 -
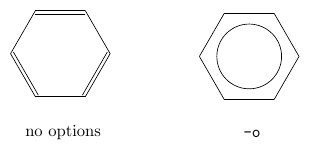
-o,--aromatic-circles(Default: False) - 芳香族の環で二重結合ではなく円で表示します。
-
-f,--fancy-bonds(Default: False) - 二重結合と三重結合をより「ファンシー」に描きます。
-
-g,--markers(Default: None) -
指定された接頭辞を使って、分子と結合にマーカーを定義します。 電子の移動を描く際などに利用できます。例えば、
-gなしでmol2chemfig -i direct CCNとすると、% 1 -[:330]% 2 -[:30,,,1]NH_2% 3が出力されますが、
mol2chemfig -g a -i direct CCNのように-gオプションを付けて実行すると、@{a1}% -[@{a1-2}:330]@{a2}% -[@{a2-3}:30,,,1]@{a3}NH_2%が得られます。
-
-n,--atom-numbers(Default: False) - 原子の番号を表示します。
-
-s,--bond-scale(Default: normalize) -
結合の長さの扱い方を指定します。
- keep: 結合の長さを変更しません。
--bond-stretchの指定は無効になります。 - normalize: 最頻出の結合長を
--bond-stretchで指定された長さにします。 - scale:
--bond-stretchで指定された値を係数として、拡大縮小します。
- keep: 結合の長さを変更しません。
-
-t,--bond-stretch(Default: 1.0) --bond-scale scaleの場合は拡大縮小の係数を、--bond-scale=normalizeの場合は最頻出の結合長を指定します。-
-w,--wrap-chemfig(Default: False) - 生成されたコードを
\chemfig{…}で囲みます。 -
-l,--submol-name(Default: None) - 名前が引数として与えられたとき、生成されたコードを
\definesubmol{name}{…}で囲みます。 -
-e,--entry-atom(Default: None) - 最初に描画される原子の番号を指定します。sub-molecule として使用される場合にのみ有効です。
-
-x,--exit-atom(Default: None) - 最後に描画される原子の番号を指定します。sub-molecule として使用される場合にのみ有効です。
-
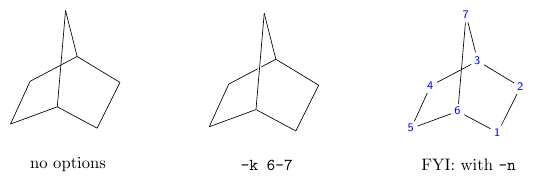
-k,--cross-bond(Default: None) - 結合が重なっている場合、手前の結合を指定して、背後の結合に切れ目を入れます。
複数ある場合は
--cross-bond 4-8,12-13のように指定します。 以下は-a 60 -w -i direct --bond-scale keep 'C1CC2CCC1C2'での-kの有無による違いです。 (この例では-kの有無で結合の長さが変わるので--bond-scale keepを付けています。) ちょっと分かりづらいですが、-kをつけたものでは交差の後ろ側の結合 3-4 が途中で切れています。
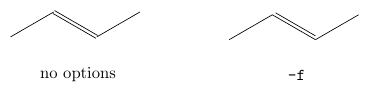
ただし、最近の Python では pip install しようとするとエラーが出るかもしれません。
少なくとも私の環境 (Arch Linux) では pip install を実行しようとすると error: externally-managed-environment
云々のエラーがでます。強制的にインストールするのであれば、以下のようにすることもできます (非推奨)。
pip install --break-system-packages --user [package]より良い方法は、仮想環境を作成しその中でインストールする方法です。一例として、以下のようにすることができます。
python -m venv mol2chemfig-env # mol2chemfig-env という名前の仮想環境を作る
. mol2chemfig-env/bin/activate # この仮想環境をアクティベート
pip install mol2chemfigPy3詳しくは Python の仮想環境等について調べてみて下さい。ここではこれ以上は深入りしません。